

DNA fragments extracted from archaeological human remains can be sequenced to identify the microorganisms that caused disease.Credit: Microgen/Getty
Over the past ten years or so, investigations of degraded or ‘ancient’ DNA have skyrocketed. By extracting DNA fragments from diverse sources —from human teeth and faeces to soil samples and ice cores — researchers have uncovered the stories of all sorts of organisms and ecosystems stretching back for millennia.
Investigators have used ancient genomics to discover the previously unknown Denisovan hominin, an evolutionary cousin and contemporary of Neanderthals that left hardly any fossil record; to identify which microorganisms caused human disease thousands of years ago; and to establish the geographical origins of domesticated plants and animals, including maize (corn) and horses1. Ancient genomics has also been used to reconstruct the composition of Pleistocene ecosystems that existed up to two million years ago2; and even to identify the wearers of ice-age jewellery3.
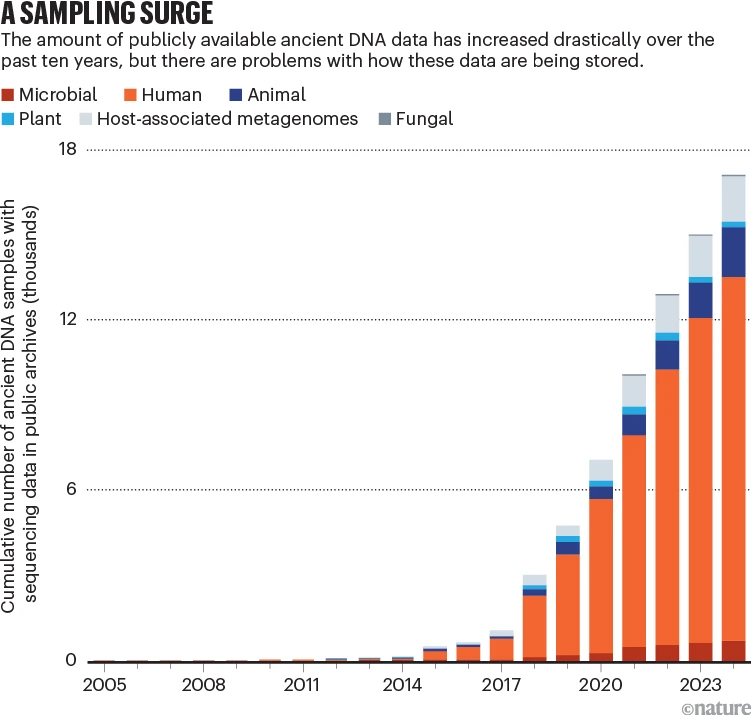
Most DNA sequence data are now archived in dedicated, publicly accessible databases, and the ancient DNA field has been heralded by some as a poster child for best practices in genetic data sharing4,5. However, as the pace of ancient DNA research has increased — largely thanks to the latest capabilities in DNA sequencing (see ‘A sampling surge’) — so, too, have problems with data archiving.
Source: https://doi.org/10.5281/zenodo.14203618. Analysis by A. Bergström et al.
Often, only some of the data obtained in any one study are uploaded to publicly available databases. Furthermore, the associated metadata — information on the age of the sample, where it was found, how the DNA was extracted and chemically treated, and so on — are frequently inaccurate or incomplete.
Here, we set out the nature of these problems and outline steps to overcome them, so that this astonishing record of the genetic past can be digitally preserved and used again and again.
Data loss
Ancient genomic data have been obtained from more than 10,000 humans6, some 700 microbes and viruses7 and, by our estimate, more than 2,000 plant and non-human animal samples. At least 2,200 ancient host-associated and environmental microbiomes (communities of microorganisms) have been sequenced7 (see also go.nature.com/3bcaxtv).
One major problem, however, is that not all sequences end up being archived.
Earlier this year, one of us (A.B.) assessed what data and metadata had been uploaded into publicly accessible databases by the authors of 42 studies of ancient DNA. All studies involved the analysis of ancient DNA extracted from humans or non-human animals, and had been published in 2021, 2022 or 2023 in the journals Nature, Science and Cell. In about half of the papers, researchers archived only those sequences that they had managed to align to a reference genome, such as that for ancient human remains, leaving no record of the unaligned sequences (see ‘A snapshot of data-archiving troubles’). This represents a permanent loss of data for more than 3,000 ancient samples analysed in just these studies8.
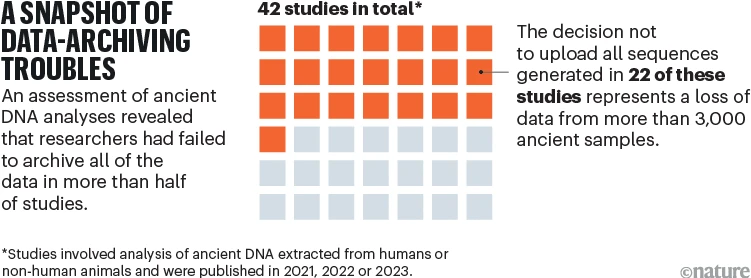
Source: Ref. 8
It might seem that any sequence that does not align to the reference genome is irrelevant. But improvements in computational methods and more-complete reference genomes could enable researchers to align such sequences in the future. Also, even if some of the unaligned sequences are not from the species of interest, this does not mean that they have no scientific value. On the contrary, these sequences could be among the most interesting in the data set, especially if they originated from pathogenic microbes that infected the host.